






LATEST
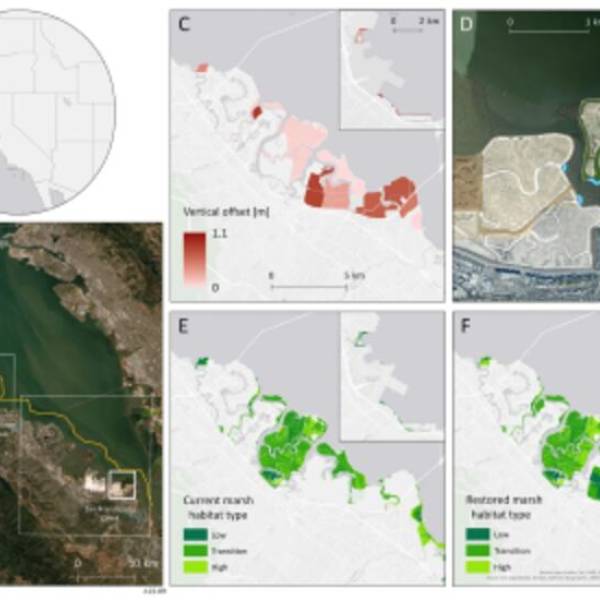
Salt marsh restoration study reveals promising...
Thu, Apr 11th 2024Supercomputer simulations demonstrate the...

Marbell leads Woolpert's geospatial business...
Wed, Apr 10th 2024Woolpert, a national architecture, engineering,...
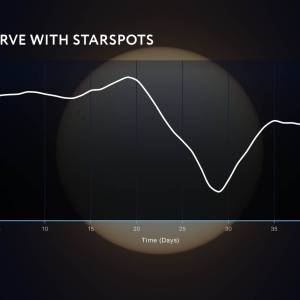
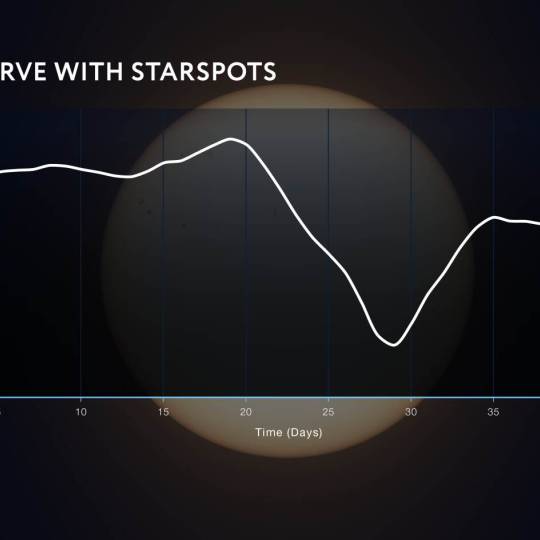
NASA's Roman Telescope uses Convolutional Neural...
Thu, Apr 4th 2024Determining the age of stars has been a...

Australia on track for unprecedented, decades-long...
Wed, Apr 3rd 2024The Australian National University (ANU) and the...
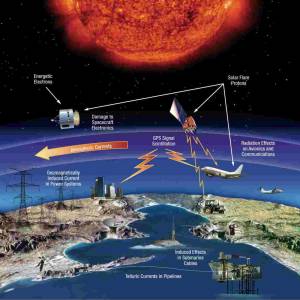
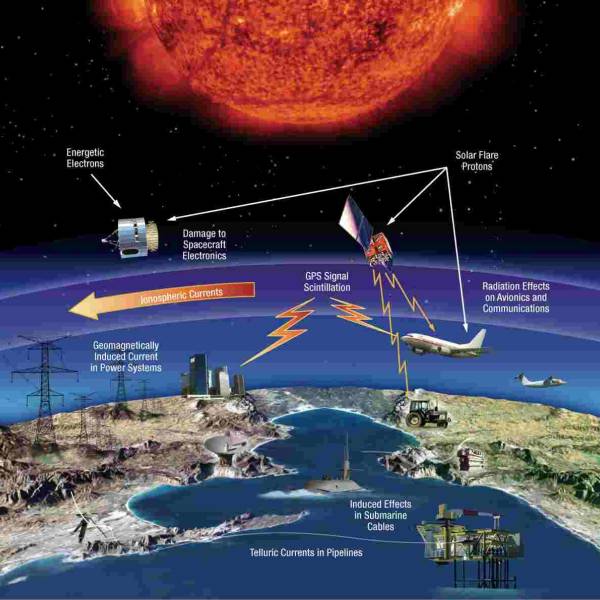
NASA predicts the Sun's corona behavior, revealing...
Tue, Apr 2nd 2024Scientists at Predictive Science are conducting a...

Revolutionizing precision agriculture: The impact...
Tue, Apr 2nd 2024The Spanish researchers in Hydraulics and...
Artificial intelligence chatbot outperforms...
Mon, Apr 1st 2024In an era of rapid technological advancements, the...

Simulations aid breakthroughs in predicting...
Thu, Mar 28th 2024Japanese scientists from Tohoku University have...

GPT-4 AI surpasses human experts in identifying...
Wed, Mar 27th 2024A recent study by researchers at Columbia...